Example
Read structure
The structure of the reads from this hypothetical sequencing technology (see image below) contains multiple regions that need to be parsed, including some of variable length.
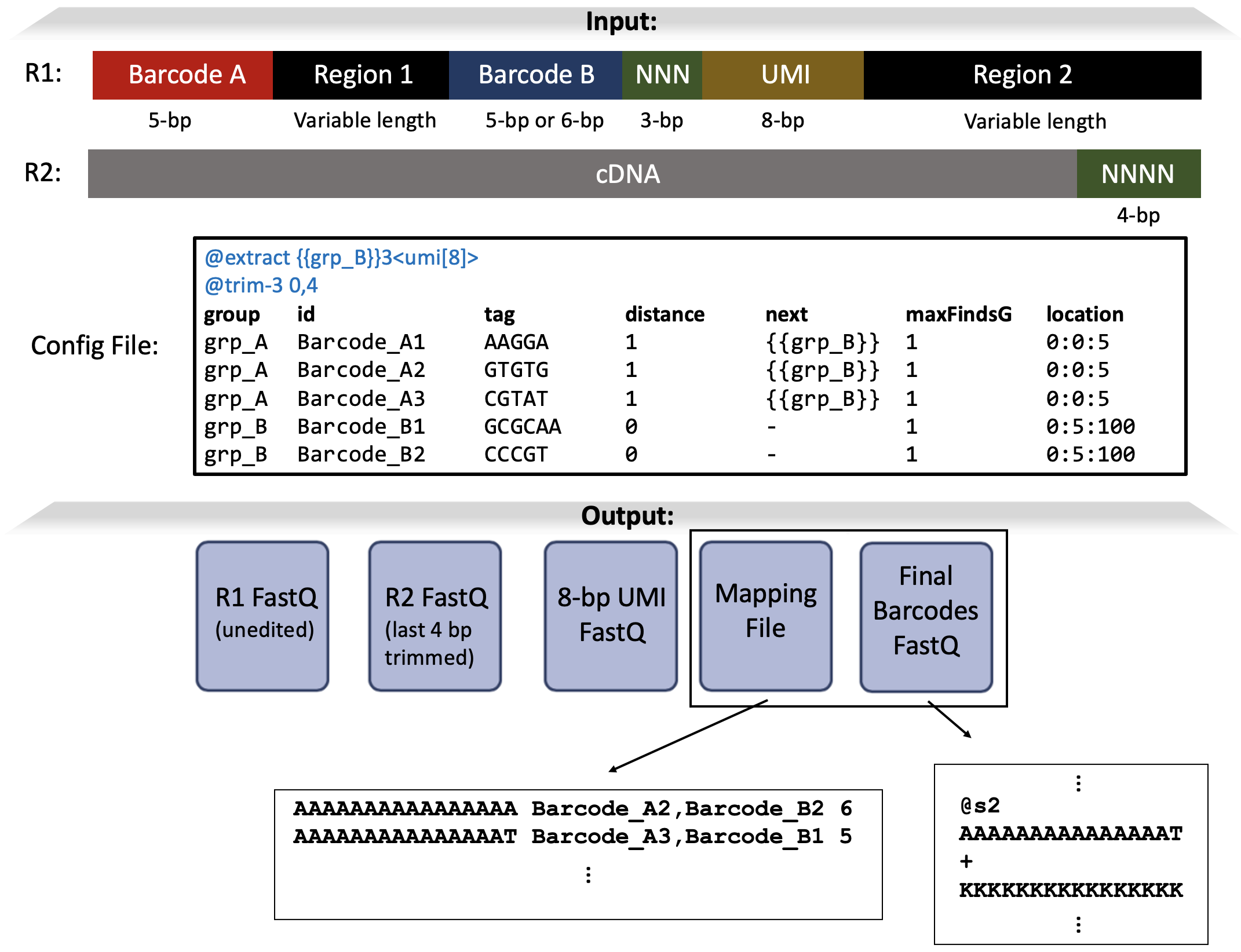
Config File
The tab-delimited table in the config file indicates the following config options:
group: Each tag belongs to a specific group. Here, the group named
grpAcontains the tags named Barcode_A1, Barcode_A2, Barcode_A3 while groupgrpBcontains the tags named Barcode_B1, Barcode_B2.id: The names of each tag. Here, there are 5 tags, one for each sequence and they are each given a unique name:
Barcode_A1,Barcode_A2,Barcode_A3,Barcode_B1,Barcode_B2tag: The sequences of the tags themselves. The tag named Barcode_A1 has sequence
AAGGA, the tag named Barcode_A2 has sequenceGTGTG, Barcode_A3 has sequenceCGTAT, Barcode_B1 has sequenceGCGCAA, and Barcode_B2 has sequenceCCCGT.distance: The tags in the grp_A group have the value
1in the distances column, meaning a hamming distance 1 error tolerance.next: The values in the next column indicate that after a grp_A tag (i.e. Barcode_A1, Barcode_A2, or Barcode_A3) is found, we should next search only for tags in the
grp_Bgroup.Note: The value grp_B is surrounded by two curly braces:
{{grp_B}}. The two curly braces indicates group. If we were searching for a tag instead of a group (like if we only wanted to search for only Barcode_B2 next), we’d use only one curly brace, i.e. {Barcode_B2}.
maxFindsG: The maxFindsG values of
1mean that the maximum number of times a specific group can be found is 1 (e.g. after finding a tag in grp_A, stop searching for tags in grp_A).location: The location for grp_A tags has the value
0:0:5, meaning that the tag is found in file #0 (i.e. the R1 file) within positions 0-5 of the read (the first 5 bp’s); for grp_B tags, splitcode searches file #0 within positions 5-100 (the next 95 bp’s).
The header in the config file indicates the following:
@extract: The expression
{{grp_B}}3<umi[8]>means that once a tag in groupgrp_Bis found in a read, splitcode will extract a sequence of length8, which we nameumi, exactly3bases after the grp_B tag is found. Thus, the grp_B tag serves as an anchor point for extracting the 8-bp sequence.Note: Just like in the “next” column, grp_B is surrounded by two curly braces:
{{grp_B}}, which indicates group. If we wanted to extract relative to a tag ID rather than a group, we’d use only one curly brace to enclose the tag ID.
@trim-3: This option specifies trimming from the 3′-end of reads. We have two values here: The first for file #0 (i.e. the R1 file) and the second for file #1 (i.e. R2 file). Thus,
0,4means we trim0bp’s (i.e. no trimming) for file #0 while we trim4bp’s off of the 3′ end of each read in file #1.
Sample Reads
Below, we’ll use four paired-end reads for demonstration purposes:
File #0: R1.fastq
@read1
GTGTCAAAAAAAAAACCCGTCCCGTGTCTCTGGGGGGGGGGGGGGG
+
CCCFFFFFHHHGGJJJJGGJJJJJJJJJJJJJJJJJJJJJIJIIGJ
@read2
AAGGAAAAAAAAAAATTTTTTTTTTTTTTTTCCCCCCCCGGGGGCG
+
CCCCFFFHHHHJGJJJJJGJJJGJJJJJJJJJJJJJJJJJJJJJJJ
@read3
GTGTGAAAAATAAAAAAACCCGTCCCGTGTCTCTGGGGGGGCCCGT
+
CCCFFFFHHHHGGGGJJGGJJJJJJJJJJJJJJJJJJJJJIJIIGJ
@read4
AAAAAAAAAAATTTTTAAAAAAATAAAAATTTAAAAAAAAAAAAAA
+
CCCFFFFHHHHGGGGJJGGJJJJJJJJJJJJJJJJJJJJJIJIIGJFile #1: R2.fastq
@read1
ATCGATATAGAGAGATACGAGAGAGAGAGATATCGAGATAGAGAGGGATTAAAAATTCCGAGACCAAAGCGCGAGCGAGAGNNCGANCGGACTTTTNAAA
+
CCCFFFFFHHHHHJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJHHHHHHFFFDD!!DDD!DDDDDDEDD!DDD
@read2
ATGGATTTAGCCCGATCCGGGTGGGAGAGATATCGAGATAGAGAGGGATATCCGGGTGGGAGAGATATATCCGGGTGGGAGAGATATGGGAGAGAGGTGG
+
CCCFFFFHHHHHHGJGJJJJJJGJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJHHHHHHFFFFDDDDDDDDDDDDDEDDDDDD
@read3
TTCGATATAGAGAGATACGAGAGAGAGAGATATCGAGATAGAGAGGGATTAAAAATTCCGAGACCAAAGCGCGAGCGAGAGGGCGACCGGACTTTTTAAA
+
CCCFFFFFHHHHHJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJHHHHHHFFFDDDDDDDDDDDDDDEDDEDDD
@read4
TATCGAGATAGAGAGGGGAGAGATATCGAGATAGAGAGGGATTAAAAATTCCGAGACCAAAGCGCGAGCGAGAGGGCGACCGGACTTTTTAAAAAAAAAA
+
CCCFFFFFHHHHHJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJHHHHHHFFFDDDDDDDDDDDDDDEDDDDDD
Command-Line Run
Given the above config file, named config.txt, the structure of our command will look as follows:
splitcode -c config.txt --nFastqs=2 --assign [output options] R1.fastq R2.fastq
The --assign option means that upon identifying the tags in reads, we’ll assign the permutation of tags to the final barcodes such that each permutation gets assigned a unique barcode.
In the next section, we will set the [output options] to specify how we want the output to be structured.
Output
Given R1.fastq, R2.fastq, and config.txt, we can specify the [output options] when running splitcode to indicate how we want to output to be structured.
Output into Separate Files
splitcode -c config.txt --nFastqs=2 --assign \
-o output_R1.fastq,output_R2.fastq --unassigned=unassigned_R1.fastq,unassigned_R2.fastq \
--outb=final_barcodes.fastq --mapping=mapping.txt \
--summary=summary.txt \
R1.fastq R2.fastq
The following output files will be generated:
output_R1.fastq and output_R2.fastq: Generated from the
-ooption, these files contain the modified versions of the original R1.fastq and R2.fastq reads. In this case, output_R2.fastq will contain the R2.fastq sequences with the last 4 bases were trimmed and the sequences within the output_R1.fastq will remain unchanged from the R1.fastq input.mapping.txt: Generated from the
--mappingoption, this file contains the mappings from the permutation of tags identified within reads to the unique final barcodes. In the right-most column of this file are numbers indicating how many times each specific mapping was found.final_barcodes.fastq: Generated from the
--outboption, this file contains the sequences of the unique final barcodes. Each of these sequences corresponds to those in output_R1.fastq and output_R2.fastq, and the mappings between these sequences and the tags are stored in mapping.txt.umi.fastq: This was generated because of the
@extract {{grp_B}}3<umi[8]>option and contains the extracted 8-bp sequences. This file is named umi.fastq because we put the name umi in the @extract string. For files in which grp_B was not identified, no extraction was performed and therefore those sequences will be blank in umi.fastq (in this case, read2).unassigned_R1.fastq and unassigned_R2.fastq: Generated from the
--unassignedoption, these files contain the reads that are considered unassigned. These sequences in these files are unmodified from the original R1.fastq and R2.fastq reads. By default, unassigned reads are those where no tag sequence could be identified (in this case, read4 is unassigned).summary.txt: Generated from the
--summaryoption, this file contains information about the splitcode run.
Now, let’s view the output files below:
@read1
AAAAAAAAAAAAAAAAGTGTCAAAAAAAAAACCCGTCCCGTGTCTCTGGGGGGGGGGGGGGG
+
KKKKKKKKKKKKKKKKCCCFFFFFHHHGGJJJJGGJJJJJJJJJJJJJJJJJJJJJIJIIGJ
@read2
AAAAAAAAAAAAAAACAAGGAAAAAAAAAAATTTTTTTTTTTTTTTTCCCCCCCCGGGGGCG
+
KKKKKKKKKKKKKKKKCCCCFFFHHHHJGJJJJJGJJJGJJJJJJJJJJJJJJJJJJJJJJJ
@read3
AAAAAAAAAAAAAAAAGTGTGAAAAATAAAAAAACCCGTCCCGTGTCTCTGGGGGGGCCCGT
+
KKKKKKKKKKKKKKKKCCCFFFFHHHHGGGGJJGGJJJJJJJJJJJJJJJJJJJJJIJIIGJ
@read1
ATCGATATAGAGAGATACGAGAGAGAGAGATATCGAGATAGAGAGGGATTAAAAATTCCGAGACCAAAGCGCGAGCGAGAGNNCGANCGGACTTTT
+
CCCFFFFFHHHHHJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJHHHHHHFFFDD!!DDD!DDDDDDEDD
@read2
ATGGATTTAGCCCGATCCGGGTGGGAGAGATATCGAGATAGAGAGGGATATCCGGGTGGGAGAGATATATCCGGGTGGGAGAGATATGGGAGAGAG
+
CCCFFFFHHHHHHGJGJJJJJJGJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJHHHHHHFFFFDDDDDDDDDDDDDEDD
@read3
TTCGATATAGAGAGATACGAGAGAGAGAGATATCGAGATAGAGAGGGATTAAAAATTCCGAGACCAAAGCGCGAGCGAGAGGGCGACCGGACTTTT
+
CCCFFFFFHHHHHJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJHHHHHHFFFDDDDDDDDDDDDDDEDD
@read1
AAAAAAAAAAAAAAAA
+
KKKKKKKKKKKKKKKK
@read2
AAAAAAAAAAAAAAAC
+
KKKKKKKKKKKKKKKK
@read3
AAAAAAAAAAAAAAAA
+
KKKKKKKKKKKKKKKK
@read1
GTGTCTCT
+
KKKKKKKK
@read2
+
@read3
GTGTCTCT
+
KKKKKKKK
@read4
AAAAAAAAAAATTTTTAAAAAAATAAAAATTTAAAAAAAAAAAAAA
+
CCCFFFFHHHHGGGGJJGGJJJJJJJJJJJJJJJJJJJJJIJIIGJ
@read4
TATCGAGATAGAGAGGGGAGAGATATCGAGATAGAGAGGGATTAAAAATTCCGAGACCAAAGCGCGAGCGAGAGGGCGACCGGACTTTTTAAAAAAAAAA
+
CCCFFFFFHHHHHJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJHHHHHHFFFDDDDDDDDDDDDDDEDDDDDD
AAAAAAAAAAAAAAAA Barcode_A2,Barcode_B2 2
AAAAAAAAAAAAAAAC Barcode_A1 1
Hint
Observe how maxFindsG works: Even though read3 has another grp_B tag, CCCGT, at the very end and it falls within the 0:5:100 location, we don’t identify it because we’ve already identified another grp_B tag earlier on the read.
There are four possibilities for identified tags in terms of groups: grp_A followed by grp_B, grp_A only, grp_B only, and no tags identified. This is thanks to the combination of the maxFindsG, next, and locations config options.
Pipe Output
Now, let’s say we want all of our output as a continuous stream written to standard output, rather than separating everything into separate files. We can do this via the --pipe option.
splitcode -c config.txt --nFastqs=2 --assign --pipe --mapping=mapping.txt R1.fastq R2.fastq
Only one file: mapping.txt will be created. Everything else will be written to standard output.
The resulting output will look as follows:
@read1
AAAAAAAAAAAAAAAA
+
KKKKKKKKKKKKKKKK
@read1
GTGTCTCT
+
KKKKKKKK
@read1
GTGTCAAAAAAAAAACCCGTCCCGTGTCTCTGGGGGGGGGGGGGGG
+
CCCFFFFFHHHGGJJJJGGJJJJJJJJJJJJJJJJJJJJJIJIIGJ
@read1
ATCGATATAGAGAGATACGAGAGAGAGAGATATCGAGATAGAGAGGGATTAAAAATTCCGAGACCAAAGCGCGAGCGAGAGNNCGANCGGACTTTT
+
CCCFFFFFHHHHHJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJHHHHHHFFFDD!!DDD!DDDDDDEDD
@read2
AAAAAAAAAAAAAAAC
+
KKKKKKKKKKKKKKKK
@read2
+
@read2
AAGGAAAAAAAAAAATTTTTTTTTTTTTTTTCCCCCCCCGGGGGCG
+
CCCCFFFHHHHJGJJJJJGJJJGJJJJJJJJJJJJJJJJJJJJJJJ
@read2
ATGGATTTAGCCCGATCCGGGTGGGAGAGATATCGAGATAGAGAGGGATATCCGGGTGGGAGAGATATATCCGGGTGGGAGAGATATGGGAGAGAG
+
CCCFFFFHHHHHHGJGJJJJJJGJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJHHHHHHFFFFDDDDDDDDDDDDDEDD
@read3
AAAAAAAAAAAAAAAA
+
KKKKKKKKKKKKKKKK
@read3
GTGTCTCT
+
KKKKKKKK
@read3
GTGTGAAAAATAAAAAAACCCGTCCCGTGTCTCTGGGGGGGCCCGT
+
CCCFFFFHHHHGGGGJJGGJJJJJJJJJJJJJJJJJJJJJIJIIGJ
@read3
TTCGATATAGAGAGATACGAGAGAGAGAGATATCGAGATAGAGAGGGATTAAAAATTCCGAGACCAAAGCGCGAGCGAGAGGGCGACCGGACTTTT
+
CCCFFFFFHHHHHJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJHHHHHHFFFDDDDDDDDDDDDDDEDD
As you can see, all the output is interleaved such that each read gets four sequences associated with it and all four sequences are outputted in order before moving on to the next read. The four sequences per read are (in order):
The unique final barcode
The extracted sequence (umi)
The output R1 sequence
The output R2 sequence
None of the unassigned reads are outputted although you can direct the unassigned reads to a file using the --unassigned option just like in the previous section.
Colab
A Google colab notebook on installing splitcode, running it on this example, and viewing its output can be found here: splitcode_example.ipynb.